Thank you to those who attended our recent webcast on Golden Helix’s SNP & Variation Suite (SVS) and its new capability related to Polygenic Risk Scores (PRS). If you were unable to attend, a recording can be found via this link. For common diseases, PRS can provide a predictive value related to the disease risk at an individual level. By… Read more »
In our recent webcast, we discussed the exciting new features of VarSeq 2.4.0 and the updated VSClinical interface. The discussion was centered around three main topics: In summary, VarSeq 2.4.0 uniquely supports the analysis of all variant types in the clinical interpretation workflow. By incorporating structural variants, enhancing automation, and empowering users to handle complex data, it offers a comprehensive… Read more »
VSClinical AMP Matching of Interpretations In this blog post, we will delve into the intricacies of the VSClinical AMP interpretation workflow. At the heart of this process lies the task of annotating cancer biomarkers with the correct interpretations based on the classification of the tumor and the type and scope of the biomarker. This is a crucial step in understanding… Read more »
As the number of genes on a gene panel increases, there is the possibility of picking up variants of medical significance that are not related to the primary indication for the test. Especially with large gene panels, exomes, and genomes, it is medically and ethically important to report variants that may be actionable to the patient. These include variants implicating… Read more »
As a lab or group scales the number of NGS samples analyzed, it is important to automate the sample analysis pipeline from the sequencer to the point where it is ready for a variant scientist or lab personnel to follow the interpretation workflow and draft a clinical report. VSPipeline leverages the core VarSeq capability to create reproducible test-specific workflows through… Read more »
Golden Helix VSClinical provides a guided workflow interface for following the ACMG and AMP guidelines to evaluate variants and CNVs for NGS tests. The output of this work is most often a lab-specific clinical report. Since it was introduced, we have provided a powerful Word-based templating system to allow labs the ability to generate customized reports to include specific content… Read more »
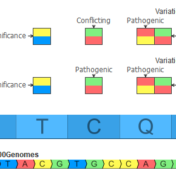
In the September 2021 monthly update to our curated ClinVar track, we made some changes that will result in roughly another 7,000 Likely Pathogenic and Pathogenic variants being available for annotation and use in the ACMG auto-classification system. Consensus Between Labs ClinVar has nearly one million unique variant classification records that are curated into multiple annotation tracks used in VarSeq and VSClinical on a monthly basis. Clinical… Read more »
One of the many tricks of encoding so much functionality into so little space in eukaryotic genomes is the ability to produce multiple distinct mRNAs (transcripts) from a single gene. While one transcript is often the dominant one for a given tissue or cell type, there are, of course, exceptions in the messy reality of biology. It doesn’t take many… Read more »
With the latest release of VarSeq, we have made significant updates to our handling of the interaction of variants and genes. This includes the support for non-coding transcripts, improved splice site predictions, and updates to gene and transcript annotations. We received several questions regarding how decisions are made in the software regarding genes and transcripts with these gene-related changes. This… Read more »
In our recent webcast announcing the upcoming release of VarSeq VSClinical and the implementation of the ACMG guidelines for NGS CNVs, we had a number of live questions we didn’t get a chance to cover at the end of the presentation. I will follow up on those questions in this blog post. But first, if you didn’t get a chance to join us for… Read more »
The potential of genetic testing to impact a patient’s life has been greatly accelerated by the sharing of variant interpretations done by clinical labs in public repositories such as ClinVar. This is not an inevitable outcome, but the persistent work and advocacy of people like Dr. Heidi Rehm and organizations like ClinGen. We recently participated in a survey and vetting… Read more »
Thank you to everyone who joined our webcast, “Whole Genome Trait Association in SVS.” If you missed the live event and are interested in knowing what we talked about, you may access the recorded event below: Our Live Q&A generated a lot of great questions. Unfortunately, we were unable to answer them all, but we have compiled some of the… Read more »
An under-appreciated area of complexity when looking into the field of genetics from the outside can be found in genes and transcripts. Alternative splicing allows eukaryotic species to have a wonderfully powerful genetic code, resulting in multiple protein isoforms being encoded in a single section of DNA. But when it comes to variant interpretation, different transcripts can result in widely different predicted… Read more »
Customizing VSClinical ACMG Guidelines Workflow Part 2 In the first part of this series, we covered how VarSeq provides customization of the clinical analysis workflow process. VSClinical’s various customization parameters within the ACMG Guidelines workflow includes the choice of how the internal knowledge base of previous variant interpretations are stored and what considerations go into this choice. In this blog, we… Read more »
Clinical labs offer a unique and sophisticated product that is performed repeatedly with high standards of quality. VarSeq was developed to provide labs with the customization required for clinical genetic tests in a repeatable workflow. On top of this, VSClinical offers additional parameters and choices that can be made when designing the test workflow. In this blog series, we will… Read more »
Genetic testing labs deal with personal data in categories with the highest level of security requirements: personal identity and medical records. Given the liability and risk associated with a breach of this secure information, it is not surprising that many labs and institutes that aggregate genomic data prefer, if not require, on-premise analysis and storage solutions. Golden Helix is in… Read more »
As clinical genetic tests have been adopted as a critical enabler of precision medicine, the number of tests offered by clinical labs and the volume of tested patients has grown by orders of magnitude in the past five years. The Gene Testing Registry, managed by the NIH, documented a rise from 13,000 to 60,000 tests offered in the US market… Read more »
If you have watched this blog over time, it would be no surprise that Golden Helix invests a lot in curating genomic annotations for use with our clinical and research analysis products. Often, we spend considerable time on the attention to detail necessary to ensure the best experience for any data source by cleaning, normalizing, documenting and then distributing it through our data annotation server. Many annotations… Read more »
VSClinical users can interpret and report genomic mutations in cancer following the AMP guidelines which we’re demonstrating in this “Following the AMP Guidelines with VSClinical” blog series. Part I introduced the hands-on analysis steps involved in creating a high-quality clinical report for targeted Next-Generation Sequencing (NGS) assays. We reviewed sample and variant quality, including the depth of coverage over the target regions by the sequencing performed for each sample. Now, we are ready to… Read more »
In the world of genomics shaping precision medicine in oncology, the limiting factor is the time-to-sign-out of a fully interpreted molecular profile report. There are many components of the entire testing process that add to the turn-around time of each test. Many of these steps, such as sample prep, sequencing, and automated secondary analysis, are bounded and consistent in their time requirements. The hands-on… Read more »