What is Genomic Prediction?
Genomic prediction is an algorithm widely used to improve desirable phenotypic traits in agriculture. For example, the cattle industry uses genomic prediction to improve beef quality and palatability as well as improve dairy production (1,2). By using genomic prediction, researchers can minimize multiple expenses in breeding industries as well as diminish the need for performing cumbersome trait data collection. To that end, we will focus on the three different methods used to run genomic prediction and how to examine the accuracy of this algorithm using K-fold cross-validation.
What are the methods for genomic prediction?
The three common methods of genomic prediction include genomic best linear unbiased prediction (gBLUP) and the Bayesian models: Bayes-C and Bayes-C-π. These methods are quite similar but differ regarding the genotypic data that is taken into consideration. Ultimately these methods estimate the effects of genetic markers in a genotyped and phenotyped training population, which are then applied to predict traits in a non-phenotyped breeding population (3).
Genomic best linear unbiased prediction (gBLUP)
The first method, gBLUP, is a linear mixed model that incorporates a marker-based genomic relationship matrix and assumes all loci contribute to the phenotype (4). This method has multiple advantages over the traditional pedigree-based models and is preferred because of its low computational demand. GBLUP output includes the Allele Substitution Effect (ASE) and the Genomic Estimated Breeding Values (EBV). The EBV can help researchers identify the plants or animals with the best breeding potential for desirable traits. This value can also identify influential loci for the phenotype of interest, which can then be used for a targeted assay for diagnostic purposes. Furthermore, since gBLUP is an all-inclusive model, SVS provides alternative large-data approaches to use memory and maximize performance efficiently.
Bayesian Methods
Bayes-C and Bayes-C- π operate differently by using a Markov Chain Montey Carlo procedure to create an ideal model based on a small set of SNPs, which differs from gBLUPs inclusion of all SNPs (4). The main difference between Bayes-C and Bayes-C- π, is that that the probability that any marker will not affect (i.e. π) remains fixed in C but is allowed to fluctuate in C- π. Similar to gBLUP, the output from these methods also include allele substitution effect, marker effect, breeding values and sample relatedness. Since not all markers are considered in the model Bayesian models are known to have more concise predictions and marker effects than gBLUP, though they are also associated with longer computing time.
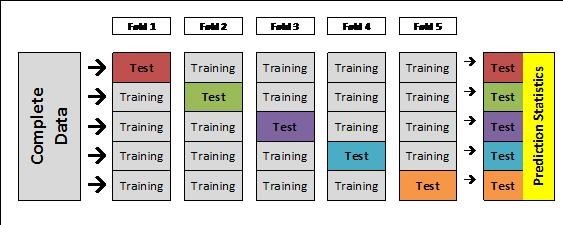
K-Fold Cross-Validation
To determine the accuracy of these genomic prediction methods, researchers can use K-fold cross-validation. The goal of cross-validation is to know how accurate your prediction dataset is by testing its ability to predict its own phenotypes when set to missing. This process divides your prediction set into several equal subsets, then runs through iterations of assigning a test set that has the phenotypes set to missing, so the remaining training sets are used for the prediction. The final step is to assess the accuracy of the predictions for the test sets, and then the user will interpret the output to assess the accuracy of their prediction dataset.
Summary
Genomic prediction is a highly valuable method for determining phenotypic traits based on genotypic information. The genomic prediction methods gBLUP, Bayes C and Bayes C-π are integrated into our SVS software and are well documented in our SVS manual. You may also want to check out our tutorials on this topic and virtual webcasts for further information. Stay tuned for more news regarding our software updates, and we hope to hear from you.
- Mateescu et al. Genetic parameters for carnitine, creatine, creatinine, carnosine, and anserine concentration in longissimus muscle and their association with palatability traits in Angus cattle. Journal of Animal Science. 2012; (12) 4238-4255.
- Gray et al. Effectiveness of genomic prediction on milk flow traits in dairy cattle. Genetics Selection Evolution. 2012; 44(1):23.
- Cericola et al. Optimizing training population size and genotyping strategy for genomic prediction using association study results and pedigree information. A case study in advanced wheat breeding lines. PLOS; 2017.
- Su et al. Comparison of genomic predictions using genomic relationship matrices built with different weighting factors to account for locus-specific variances. Journal of Dairy Science. 2014; (10) 6547-6559.