With the recent upgrade to VarSeq 1.4.7, users gain access to some new great features. Among the additions are new CNV annotations (Figure 1).
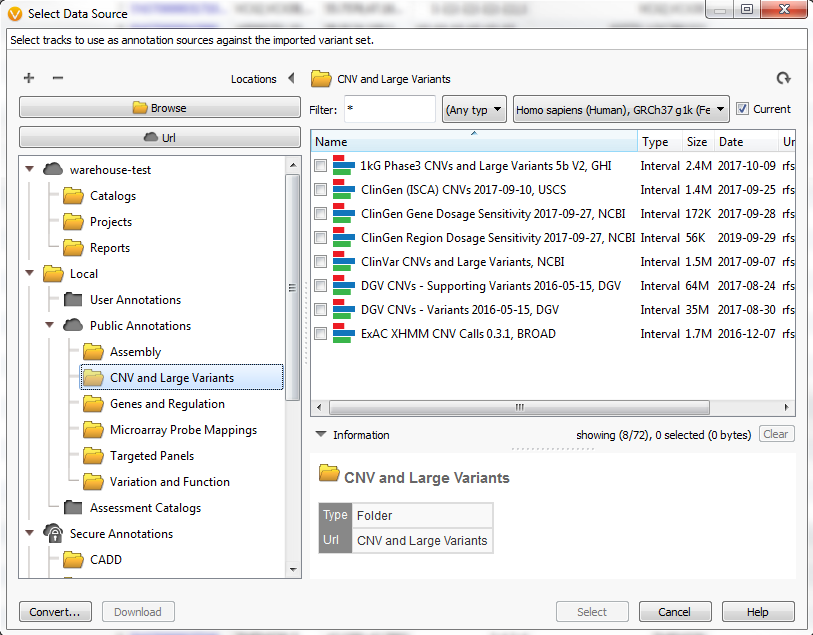
Fig 1. The new directory of Golden Helix curated annotations dedicated to CNVs.
In this final chapter of the annotation blog series, we are going to provide descriptions of the new CNV annotations and how they can be used. The types of CNV annotations vary and include frequency, clinical assessment, and research evidence tracks. A possible first step in any variant analysis may be to capture those rarely found in the population.
1000 Genome Phase3 CNVs and Large Variants
Copy number variants in 1kG Phase3 CNVs and Large Variants (>45,000 variants) are collected from the same 2504 samples used for the variant frequency annotation. It contains descriptions of CNVs, allele frequencies, allele counts, heterozygous and homozygous counts in total and for each population. When loading 1kG CNVs into your VarSeq project, you’ll see two sources in the CNV table (Figure 2); Summary of CNVs and Overlapping CNVs.
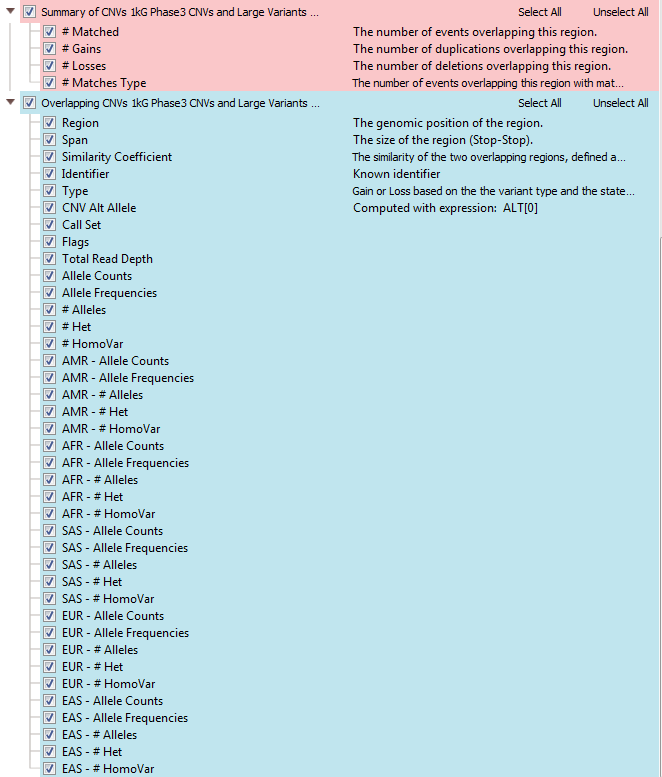
Fig 2. 1kGenome Phase 3 Summary and Overlapping CNV fields shown in VarSeq.
Every CNV annotation will have a similar format, with a summary and overlapping CNV tables. The summary table lists how many regions the called CNV matches in 1kG Phase3, whether it is gain or loss and how many match that loss/gain type. The frequencies for each subpopulation are also available for CNVs, and here is a breakdown of each abbreviation:
- AMR-Latino (mixed American)
- AFR-African
- SAS-South Asian
- EUR-European
- EAS-Eastern Asian
In addition to 1kGenome CNV population frequency information, Golden Helix has also curated ExAC CNVs to add more to the population-based CNV interpretation.
ExAC XHMM CNV
The Exome Aggregation Consortium utilized XHMM for calling CNVs, and thus Golden Helix has curated the ExAC XHMM CNV Calls. ExAC CNVs provides a PHRED score representing the confidence of the XHMM call and can be used to filter your CNVs. This dataset spans 61,486 unrelated individuals and a CNV event from each individual represented independently. Again, ExAC has two source field groups; Summary of CNVs and Overlapping CNVs.
The Overlapping CNVs fields
- Region and Span (Size): Genomic regions & total number of base pairs for annotated CNV
- Similarity Coefficient: % overlap of CNV to annotated CNV
- Type: type of CNV event
- Population: individual with CNV’s associated population
- Quality Score: Phred Scaled Likelihood Score
The coupling of frequency data in both ExAC and 1kGenome can serve as powerful tools to isolate rarely occurring CNVs in multiple populations. The next step in your CNV analysis may be to determine any clinical relevance.
ClinGen
The Clinical Genome Resource is an NIH funded program providing research and clinical genetic testing information from the National Center for Biotechnology Information (NCBI) and the National Library of Medicine (NLM). There are three ClinGen tracks available; Region Dosage Sensitivity (>60 variant regions), Gene Dosage Sensitivity (>1,200 variants), and Overlapping CNVs (>36,000 variants). You will notice that the region and gene dosage sensitivity tracks have similar fields (Figure 3). However, the differences are ‘region’ based data and ‘gene’ based data within a region. ClinGen also provides Haploinsufficiency (HI) and Triplosensitivity (TS) data. The haploinsufficiency score should be used to evaluate deletions and Triplosensitivity scores for duplications. The HI and TS scores is assessed with additional evidence supporting the pathogenicity score and links to reference information (Figure 3).


Fig 3. Region and Gene Dosage fields from the ClinGen annotation track; containing the Haploinsufficiency and Triplosensitivity scores.
ClinGen’s Overlapping CNVs track (Figure 4) provides a variety of unique fields with specific sample/study details including study conditions, associated phenotypes, and additional database IDs.
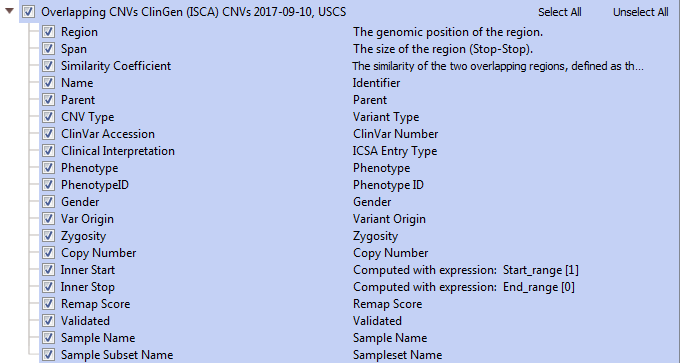
Fig 4. List of fields from the ClinGen (ISCA) CNV annotation from VarSeq.
Additional descriptions of the fields in Figure 4:
- Name and Parent: NCBI’s IDs for samples and sample’s parent
- CNV type: Loss or Gain
- ClinVar Accession and Clinical Interpretation: ClinVar ID and pathogenicity
- Benign
- Missing
- Likely Benign
- Uncertain
- Pathogenic
- Likely Pathogenic
- Phenotype and ID: Human Phenotype Ontology listed phenotypes and IDs
- Gender, Var Origin, Zygosity: details of alleles origin
- Copy Number: copy number associated with loss or gain
- Inner Start/Stop: positions of each start and stop region for each CNV
- Remap Score: NCBI’s Remap score for multiple assemblies
- Score around 1: region relatively unchanged between assemblies
- >1: insertion in target assembly
- <1: deletion in target assembly
- Validated: CNV calls validated with additional methods
- Sample Name and Sample Subset Name: describes any division of samples in the study
VarSeq’s GenomeBrowse provides a simple solution to aid in CNV analysis. Figure 5 shows GenomeBrowse with the RD CRExome-1 sample Duplicate CNV plotted below ClinGen’s overlapping CNVs. Also, in Figure 5 is VarSeq’s Console view which is accessed by clicking on any plotted element in GenomeBrowse. This console is similar to VarSeq’s ‘Details View’ providing a useful approach to interpret each field in ClinGen with data associated to a specific CNV.
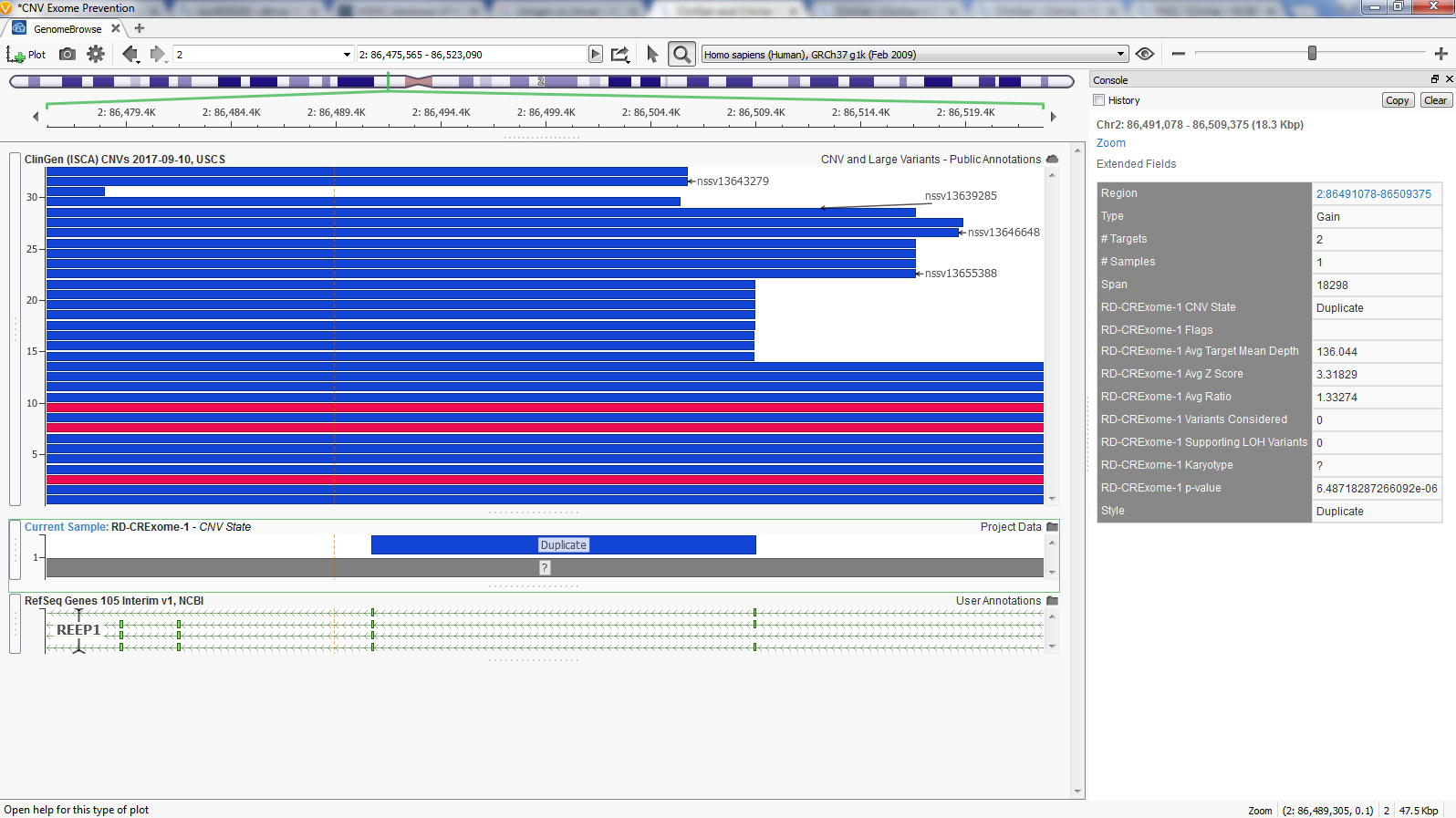
Fig 5. ClinGen and VarSeq sample CNVs plotted in GenomeBrowse with additional CNV information in the console view.
The effort behind ClinGen’s goal to create an authoritative central resource for clinical assessment of genes and variants is tightly linked to NCBI’s ClinVar, and we provide both as separate annotations in VarSeq.
ClinVar CNVs and Large Variants
ClinVar CNVs and Large Variants (>21,000 variants) contain all variants in the ClinVar database that are over 200 base pairs long. CNV information coming from ClinVar is also linked to NCBI’s dbVar database. Within ClinVar, there is access to multiple IDs (allele, gene, HGNC, dbVar nsv/esv, MedGen phenotypeIDs, and others), study origin, and clinical significance. Overall, ClinVar provides data about phenotypes and supporting evidence for variants. Figure 6 is of VarSeq’s Detail View that you can access by clicking on any variant in the Table View. This Details View is an efficient way to view the abundant information found in ClinVar, while also providing the hyperlink access to NCBI sites.
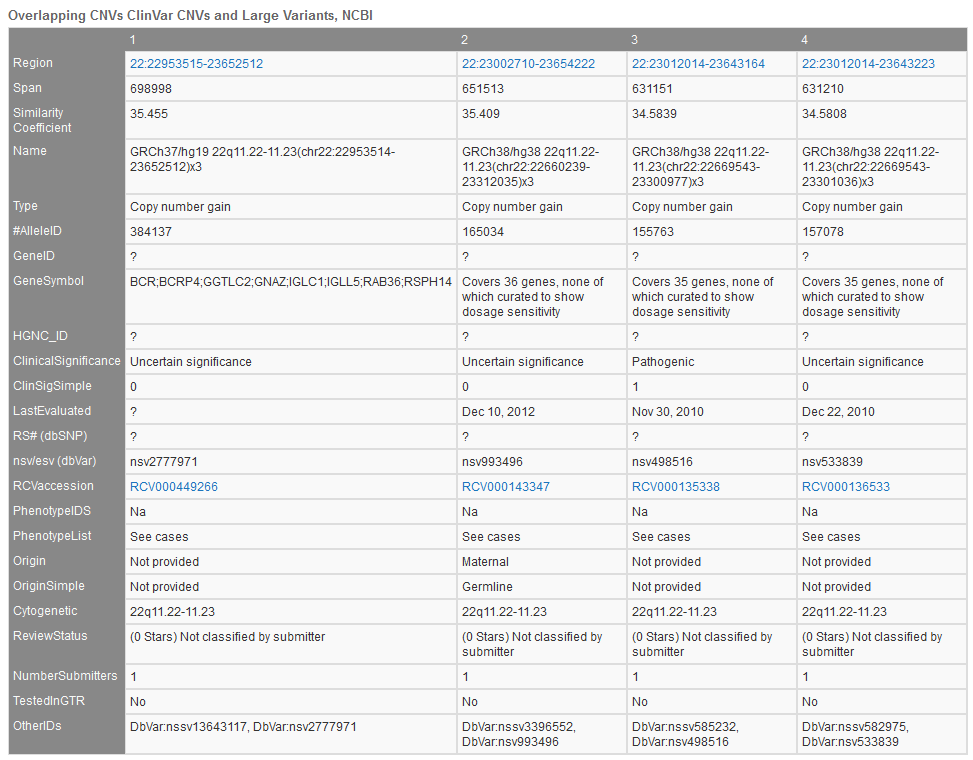
Fig 6. ClinVar fields and data presented in VarSeq’s ‘Detail View’.
Though ClinVar provides supporting evidence for variants, another annotation track in VarSeq delivers a vast increase in the number of variants with associated research information.
DGV CNVs
The Database of Genomic Variants seeks to provide a comprehensive summary of structural variation in the human genome. This database includes not only CNVs (greater than 1000 bp) but also insertions/deletions (InDels) (as low as 100 bp), and inversions/inversion breakpoints. The variants in this database were identified in healthy control samples only. The information within DGV is presented with two separate tracks in VarSeq; Supporting Variants (>6 million variants) and Variants (>300,000 variants). Both tracks supply similar cohort/study information and hyperlinks for fields such as PubMedIDs, method descriptions, and cohort descriptions (Figure 7). The main difference between DGV Variants and Supporting Variants is the depth of information. Seen in Figure 8, for the same five CNVs called in VarSeq, the number of matched variants for the Supporting Variants track is much greater. The Variants track supplies a compressed or summarized version of the same study information.

Fig 7. Fields in the DGV CNVs – Variants annotation showing study details.
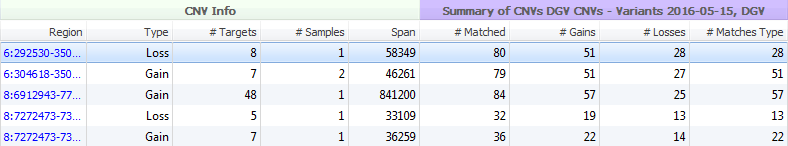
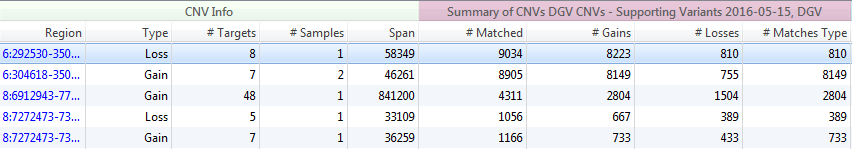
Fig 8. A Comparison between DGVs Supporting Variants and Variants. Supporting Variants cohort information is much more expansive, while the Variants provides some summarized research details.
Hopefully, this blog series has provided clarity on the types of annotations curated by Golden Helix. Again, it is important to mention our effort behind data curation. Our team is keen on formatting these annotations, suitably providing all the necessary information for efficient variant analysis. We would encourage you to explore each of these annotations to learn even more about the information contained and its source. As always, customers of Golden Helix have full access to support and we will assist you with any questions you have during your annotation exploration.