Meta-analysis is an important tool to have in the bioinformatics toolbox. The numbers alone speak for themselves. It is the fourth most requested feature for SVS, and a simple google scholar search for 2014 and 2015 find 17,300 results for genetics + meta-analysis. There are several meta-analysis utilities out there that will take results from studies and perform the meta-analysis…. Read more »
As VarSeq has been evaluated and chosen by more and more clinical labs, I have come to respect how unique each lab’s analytical use cases are. Different labs may specialize in cancer therapy management, specific hereditary disorders, focused gene panels or whole exomes. Some may expect to spend just minutes validating the analytics and the presence or absence of well-characterized… Read more »
Personal genome sequencing is rapidly changing the landscape of clinical genetics. With this development also comes a new set of challenges. For example, every sequenced exome presents the clinical geneticist with thousands of variants. The job at hand is to find out which one might be responsible for the person’s illness. In order to reduce the search space, clinicians use various methods… Read more »
Last month, Dr. Bryce Christensen presented Population-Based DNA Variant Analysis via webcast. The webcast reviewed the fundamentals of population-based variant analysis and demonstrated some of the tools available in SVS for analysis of both common and rare variants such as the SKAT-O method, as well as other functions for annotation, visualization, quality control and statistical analysis of DNA sequence variants. Here… Read more »
Our Genomic Prediction webcast in December discussed using Bayes-C pi and Genomic Best Linear Unbiased Predictors (GBLUP) to predict phenotypic traits from genotypes in order to identify the plants or animals with the best breeding potential for desirable traits. The webcast generated a lot of good questions as our webcasts generally do. I decided to begin to share these Q&A… Read more »
Genotype imputation is a statistical technique for estimating sample genotypes at loci that were not directly assayed by sequencing or microarray experiments. There are several reasons why you might want to use imputation in a research study. For example: Improve call rates in GWAS by imputing sporadic missing genotypes Harmonize the data content from different GWAS genotyping platforms so that… Read more »
In 1914 the German cytologist Theodor Boveri coined the phrase “Cancer is a disease of the genome”. At this time his ideas were equally revolutionary as they were highly contested. Fast forward. More than hundred years later, Next-Generation Sequencing effectively permits a highly sensitive analysis of cancer cells. It can help us to understand mutations associated with cancer development and… Read more »
The Integrative Therapies Institute is soon hosting the annual, ITI 2015 conference January 23rd through the 25th in sunny San Diego and our own Dr. Andreas Scherer has been invited to speak. Some of the most prominent genomic and integrative medicine specialists will gather at ITI 2015 to share case studies and protocols with the community. Attendees can expect to… Read more »
If you haven’t been closely watching the twittersphere or other headline sources of the genetics community, you may have missed the recent chatter about the whole genome sequencing of 17 supercentenarians (people who live beyond 110 years). While genetics only explains 20-30% of the longevity of those with average life-spans, it turns out there is a number of good reasons… Read more »
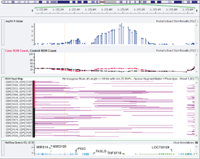
It’s come to my attention in recent weeks, through various customer interactions, that many are not aware of the fantastic functionalities that exist in SNP and Variation Suite (SVS) for large-n DNASeq workflows; this includes large cohort analyses with case/control variables. The data you’ll see below is the publically available 1kG Phase 1 v3 Exome sequences from 1,092 individuals with… Read more »
Golden Helix is proud to announce the release of the Golden Helix GenomeBrowse Plugin for Ion Torrent server. The new plug-in enables adding selected BAM files from Torrent Server reports directly into GenomeBrowse. The BAM files remain on the torrent server and are streamed from the server on demand using your credentials. This feature allows GenomeBrowse users to visualize genomic… Read more »
Recently, I have been thinking a lot about Human Genome Variation Society (HGVS) notation — you know “G dot”, “P dot”, and “C dot”. HGVS has quickly become one of the most common ways to represent variants. It’s no wonder that HGVS nomenclature is used so widely. It provides an easily readable, compact representation of a variant. Since it is… Read more »
Tutorials are ever-present in the world today, and for good reason. Why struggle through a complicated process yourself, when there is already a guide established to assist? While no one would suggest that a tutorial is the only way to complete a project, it is certainly a nice starting point. This rings true with genetic software as well. There are… Read more »
Genomic research is exploding. There is a plethora of new methods and workflows for research and clinical use. While we are a software company at heart, we find ourselves in the role of educators. Our customer interactions are about informing, teaching, and consulting. A few years back, we started with regular webcasts that took this idea to the next level…. Read more »
Over the last decade, DNA sequencing has made vast technological improvements. With the cost of sequencing decreasing significantly, sequencing technology has become a product for the masses. The sequencing technology and programs that were once used exclusively by major research institutions are now becoming available in many research facilities around the globe. These tools produce large amounts of data sets… Read more »
You probably haven’t spent much time thinking about how we represent genes in a genomic reference sequence context. And by genes, I really mean transcripts since genes are just a collection of transcripts that produce the same product. But in fact, there is more complexity here than you ever really wanted to know about. Andrew Jesaitis covered some of this… Read more »
For the SVS 8.2 release we decided to improve upon the existing ROH feature. The improvements include new parameters to define a run and a new clustering algorithm to aide in finding more stringent clusters of runs. The improvements were motivated by customer comments and a recent research paper by Zhang 2013, “cgaTOH: Extended Approach for Identifying Tracts of Homozygosity,”… Read more »
“Who has ever had a bad experience with a VCF file?” I like to ask that question to the audience when I present data analysis workshops for Golden Helix. The question invariably draws laughter as many people raise their hands in the affirmative. It seems that just about everybody who has ever worked VCF files has encountered some sort of… Read more »
Up until a few weeks ago, I thought variant classification was basically a solved problem. I mean, how hard can it be? We look at variants all the time and say things like, “Well that one is probably not too detrimental since it’s a 3 base insertion, but this frameshift is worth looking into.” What we fail to recognize is… Read more »
On my flight back from this year’s Molecular Tri-Conference in San Francisco, I couldn’t help but ruminate over the intriguing talks, engaging round table discussions, and fabulous dinners with fellow speakers. And I kept returning to the topic of how we aggregate, share, and update data in the interest of understanding our genomes. Of course, there were many examples of… Read more »