An Example of an Integrated Clinical Workflow for CNVs and SNVs
In this blog series, I discuss the architecture of a state of the art secondary pipeline that is able to detect single nucleotide variations (SNVs) and copy number variations (CNVs) in one test leveraging next-gen sequencing. In Part I, we reviewed genetic variation in humans and looked at the key components of a systems architecture supporting this kind of analysis. Part II reviews how algorithms such as GATK are leveraged to call single nucleotide variations. Part III will give you an overview of some of the design principles of a CNV analytics framework for next-gen sequencing data. Part IV shows some examples of how a CNV caller identifies CNVs. Finally, Part V shows what an integrated clinical workflow looks like.
Let’s start with a specific case. A patient’s DNA was sequenced using the TruSight Cardio Sequencing Kit from Illumina to identify causal variants implicated in inherited cardiac conditions (ICCs). This NGS gene panel test resulted in over 2,000 variants in 174 genes with known associations to 17 ICCs. To evaluate variants of clinical relevance, we must develop a workflow that:
- Identifies mutations in regions targeted by the gene panel
- Filters out low quality variants
- Identifies variants that are classified as pathogenic with a predicted missense or loss of function effect
The filtering procedures defined in the above template can be seen in figure 1.
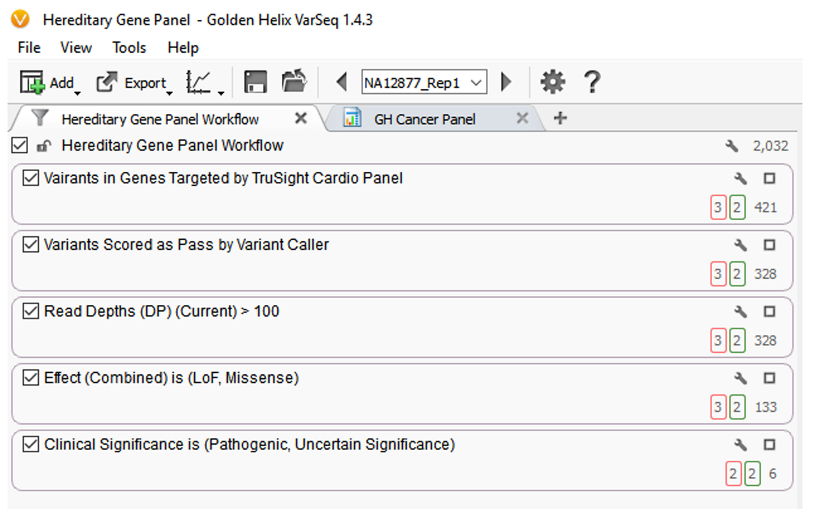
Fig 1: VarSeq filters and Hereditary Panel Workflows
This first filter card removes variants that are present outside of the regions sequenced in the gene panel. Variants that exist outside of gene regions targeted by the panel are not the focus of this test and are likely not covered with sufficient reads to be high quality, and thus should be removed. The 421 variants remaining are then passed through a second filter which identifies which variants have a classification of “PASS” as determined by the variant caller. Also, using data from the VCF file created by the variant caller, variants are filtered (filter card 3) to keep those with a read depth greater than or equal to the specified threshold of 100. This is an example of a value tuned during the test validation to ensure the desired trade-off between sensitivity and specificity. These filtering steps produce a list of 328 variants.
The next step in the filter chain is to categorize variants by sequence ontology effect prediction, and only those variants that present a loss of function or missense are included, resulting in 133 variants. Finally, to keep only variants of clinical relevance, a filter on whether a variant is annotated in the ClinVar database as “Pathogenic” or Likely Pathogenic” is applied. This reduces the number of variants down to 6.
CNV Analysis
Copy Number Variants (CNVs) provide critical evidence for many genetic tests run in a clinical lab. Along with the small variants, NGS data can also be used to call CNVs, providing extra value for data you may already have and discovering events that may not be captured by any of your existing testing technology.
The CNV algorithm implemented in VarSeq uses sample-level coverage statistics to detect CNV events. The coverage data for a given region is normalized against the same region in closely matched controls, and then metrics from this comparison (Z-score and Ratio) can be used to call single or double copy losses (deletion) or copy gain (duplication) CNV events.
The Z-score for a target measures the number of standard deviations a sample’s coverage is from the mean reference sample coverage, while the Ratio is the target coverage divided by the mean reference sample coverage.
In our example, the patient DNA was sequenced using the TruSight Cardio Sequencing kit. Variant filtration and annotation in VarSeq resulted in 6 variants classified as “Pathogenic” in the ClinVar database. In addition to the 6 pathogenic variants… to continue reading, I invite you to download a complimentary copy of my eBook. You can do so by clicking the button below.