Examples of CNV Calling
What do CNV calls actually look like? What are some of the key metrics to determine an event? Part IV of the Secondary Analysis 2.0 blog series will answer these questions by walking through some examples of how our CNV caller, VS-CNV, identifies CNVs.
Golden Helix integrates multiple metrics to determine if a CNV event is present. These metrics are:
- Z-score: The Z-score measures the number of standard deviations a target is from the reference sample mean. It is computed by subtracting the normalized read depth of the reference samples from the normalized depth for the sample of interest and dividing the result by the standard deviation of the reference samples. A high Z-score is indicative of a duplication event, while a lower Z-score is evidence for a deletion event. The Z-scores are also used to compute p-values for each called event. The p-value for an event measures the probability of Z-scores at least as extreme assuming the event targets are diploid and can be useful for evaluating call quality.
- Ratio: The ratio is computed for a given target by dividing the normalized read depth for the sample of interest by the normalized mean depth over the reference samples. If no CNV event is present, the sample of interest should have the same normalized depth as the reference samples, indicating a ratio value close to 1, while homozygous deletions, heterozygous deletions, and duplications will have ratio values around 0, 0.5 and 1.5, respectively. Unlike the Z-score, the ratio gives us the ability to differentiate between homozygous, and heterozygous deletion events.
- Variant allele frequency (VAF).
The first two metrics are computed from normalized coverage and provide the primary evidence used to identify CNV events.
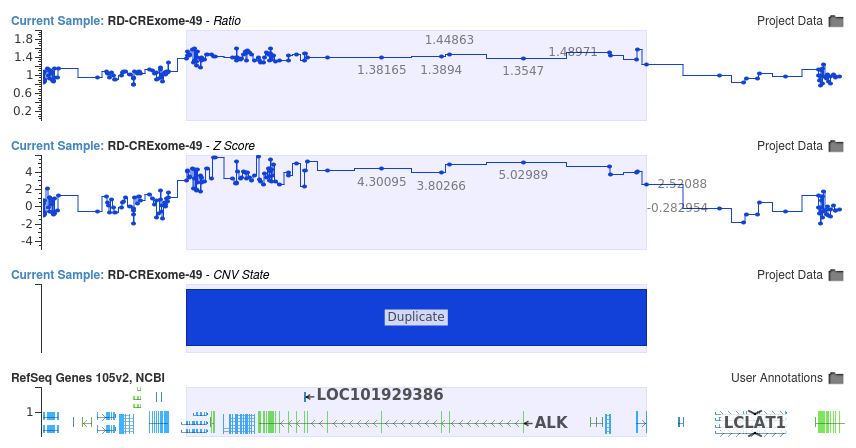
Fig 1: Multi-Gene Duplication
The combination of the Z-score and ratio allows us to detect CNV events ranging from small single exon events to large whole chromosome events. Figure 1 shows a large multi-gene duplication event, encompassing the ALK gene. The large Z-score indicates that targets within this event are around 5 standard deviations from the reference samples. These large Z-scores, combined with the ratio values centering around 1.5, provide… to continue reading, I invite you to download a complimentary copy of my eBook. You can do so by clicking the button below… to continue reading, I invite you to download a complimentary copy of my eBook. You can do so by clicking the button below.