Personal genome sequencing is rapidly changing the landscape of clinical genetics. With this development also comes a new set of challenges. For example, every sequenced exome presents the clinical geneticist with thousands of variants. The job at hand is to find out which one might be responsible for the person’s illness.
In order to reduce the search space, clinicians use various methods to filter out noise. Case-cohort analysis or sequencing additional family members can also improve diagnostic accuracy by eliminating variants that are present in non-carriers that are also present in the cases. There have been a vast amount of algorithms and filters developed for those scenarios.
Unfortunately, clinicians mostly deal with affected individuals and/or small families. Conventional whole-genome and whole-exome search and variant-prioritization tools are under powered in these situations, potentially limiting the number of successful diagnoses. In order to further reduce the search space and to focus the analysis towards variant candidates that with high likelihood are impacting the observed symptoms, we have chosen to implement a phenotype driven variant ontological re-ranking tool in VarSeq called PhoRank.
PhoRank is modeled on the Phevor algorithm [1] that was published by Mark Yandell’s group in 2014. It works by leveraging the knowledge resident in diverse biomedical ontologies, such as the Human Phenotype Ontology (HPO), and the Gene Ontology (GO). These Ontologies contain the critical links between gene and disease associations. The Ontologies are organized as directed acyclic graphs, allowing different traversals to take you from one input term, through connections, to other term nodes and their connections.
PhoRank and Phevor start by inputting individual’s phenotypes to well specified terms in the Human Phenotype Ontology. It assigns initial scores to these nodes in the ontology graph. Next the algorithm propagates this score information through the various ontologies, with the score decaying the further they are from the original nodes. When finished, the genes observed in the ontologies with high scores are more closely related to the specified phenotypes, while genes with low scores have little or no relation to the phenotypes.
VarSeq then takes these ranked genes and joins them to the genes observed in the imported and annotated variants from your project. The output contains the gene score, a useful percentile ranking of that gene amongst all other observed, as well as an informative path between the gene and the closest input phenotype term.
The result is VarSeq can harmonize ranking and filtering strategies, allowing some filtering to narrow the search space, while prioritizing the resulting variants by their gene’s relevance to an individual’s phenotypes.
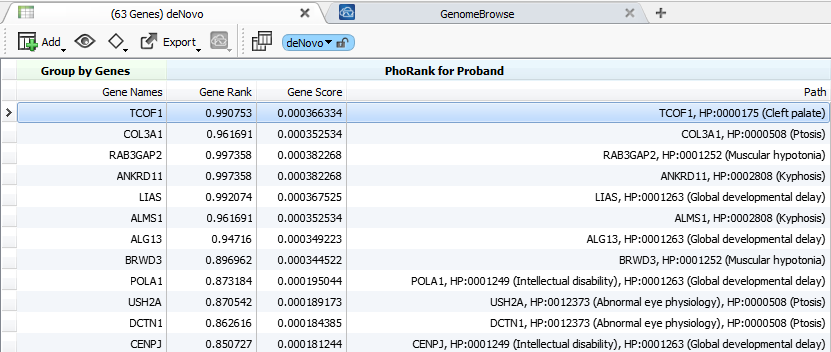
PhoRank results showing prioritized candidate genes with de Novo mutations in a proband with global developmental delay, cleft palate, and a few other phenotypes.
PhoRank is especially useful for single-exome and family-trio-based diagnostic analyses, the most commonly occurring clinical scenarios. Please check out our newest release of VarSeq for more information. Our team of experts are happy to demonstrate how you can use this latest addition to VarSeq’s capabilities to conduct a whole exome analysis more effectively .
Literature
[1] Phevor Combines Multiple Biomedical Ontologies for Accurate Identification of Disease-Causing Alleles in Single Individuals and Small Nuclear Families. Am J Hum Genet. 2014 Apr 3;94(4):599-610